
![]() CompBioMed is an EU-funded project working on applications for computational biomedicine. It is part of a vast international consortium across Europe and USA working on urgent coronavirus research. “Modelling and simulation is being used in all aspects of medical and strategic actions in our fight against coronavirus. In our case, it is being harnessed to narrow down drug targets from billions of candidate molecules to a handful that can be clinically trialled”, says Peter Coveney from University College London (UCL) who is heading CompBioMed’s efforts in this collaboration. The goal is to accelerate the development of antiviral drugs by modelling proteins that play critical roles in the virus life cycle in order to identify promising drug targets.
CompBioMed is an EU-funded project working on applications for computational biomedicine. It is part of a vast international consortium across Europe and USA working on urgent coronavirus research. “Modelling and simulation is being used in all aspects of medical and strategic actions in our fight against coronavirus. In our case, it is being harnessed to narrow down drug targets from billions of candidate molecules to a handful that can be clinically trialled”, says Peter Coveney from University College London (UCL) who is heading CompBioMed’s efforts in this collaboration. The goal is to accelerate the development of antiviral drugs by modelling proteins that play critical roles in the virus life cycle in order to identify promising drug targets.
Secondly, for drug candidates already being used and trialled, the CompBioMed scientists are modelling and analysing the toxic effects that these drugs may have on the heart, using supercomputing resources required to run simulations on such scales. The goal is to assess the drug dosage and potential interactions between drugs to provide guidance for their use in the clinic.
Finally, the project partners analysed a model used to inform the UK Government’s response to the pandemic. It has been found to contain a large degree of uncertainty in its predictions, leading it to seriously underestimate the first wave. “Epidemiological modelling has been and continues to be used for policy-making by governments to determine healthcare interventions”, says Coveney. “We have investigated the reliability of such models using HPC methods required to truly understand the uncertainty and sensitivity of these models.” As a conclusion, a better public understanding of the inherent uncertainty of models predicting COVID-19 mortality rates is necessary, saying they should be regarded as “probabilistic” rather than being relied upon to produce a particular and specific outcome.

BioExcel![]() is an EU-funded project developing some of the most popular applications for modelling and simulations of biomolecular systems. Along with code development, the project builds training programmes to address competence gaps in extreme-scale scientific computing for beginners, advanced users and system maintainers.
is an EU-funded project developing some of the most popular applications for modelling and simulations of biomolecular systems. Along with code development, the project builds training programmes to address competence gaps in extreme-scale scientific computing for beginners, advanced users and system maintainers.
When COVID-19 struck, BioExcel launched a series of actions to support the community on SARS-CoV-2 research, with an extensive focus on facilitating collaborations, user support, and providing access to HPC resources at partner centers. BioExcel partnered with Molecular Sciences Software Institute to establish the COVID-19 Molecular Structure and Therapeutics Hub to allow researchers to deposit their data and review other group’s submissions as well.
During this period, there was an urgent demand for diagnostics and sharing of data for COVID-19 applications had become vital more than ever. A dedicated BioExcel-CV19 web-server interface was launched to provide access to study molecules involved in the COVID-19 disease. This allowed the project to be a part of open access initiative promoted by the scientific community to make research accessible.
Recently, BioExcel endorsed the EU manifesto for COVID-19 Research launched by European Commission as part of their response to the coronavirus outbreak.
![]() MaX (MAterials design at the eXascale) is a European Centre of Excellence aiming at materials modelling, simulations, discovery and design on the exascale supercomputing architectures.
MaX (MAterials design at the eXascale) is a European Centre of Excellence aiming at materials modelling, simulations, discovery and design on the exascale supercomputing architectures.
Though the main interest of the MaX flagship codes is then centered on materials science, the CoE is participating in the fight against SARS-CoV-2. Given the critical pandemic situation that the world is currently facing, an unprecedented effort is being devoted to the study of SARS-CoV-2 by researchers from different scientific communities and groups worldwide. From the biomolecular standpoint, particular focus is being devoted to the main protease, as well as to the spike protein. As such, it is an important potential antiviral drug target: if its function is inhibited, the virus remains immature and non-infectious. Using fragment-based screening, researchers have identified a number of small compounds that bind to the active site of the protease and can be used as a starting point for the development of protease inhibitors.
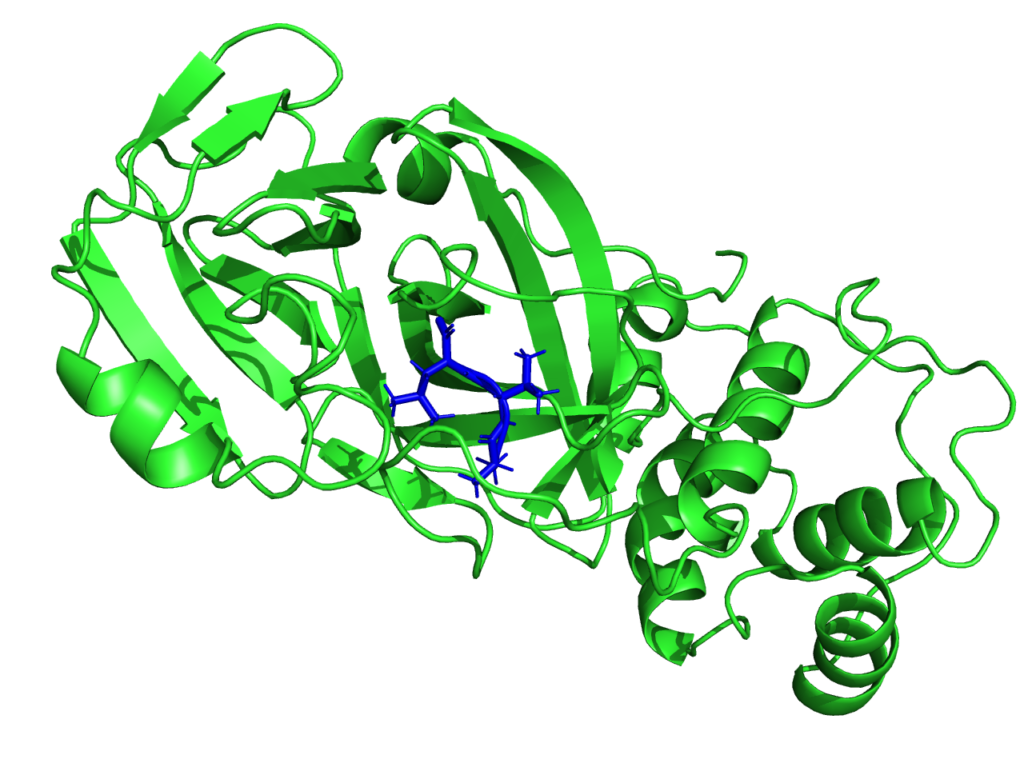
Among other quantities, MaX researchers now have the possibility to model the electronic structure of the protease in contact with a potential docked inhibitor, and provide new insights on the interactions between them by selecting specific amino-acids that are involved in the interaction and characterizing their polarities. This new approach proposed by the MaX scientists is complementary to the docking methods used up to now and based on in-silico research of the inhibitor. Biological systems are naturally composed of fragments such as amino-acids in proteins or nitrogenous bases in DNA.
With this approach, it is possible to evaluate whether the amino acid-based fragmentation is consistent with the electronic structure resulting from the QM computation. This is an important indicator for the end-user, as it enables to evaluate the quality of the information associated with a given fragment. Then, QM observables on the system’s fragments can be obtained, which are based on a population analysis of electronic density of the system, projected on the amino-acid.
A novelty that this approach enables is the possibility of quantifying the strength of the chemical interaction between the different fragments. It is possible to select a target region and identify which fragments of the systems interact with this region by sharing electrons with it.
“We can reconstruct the fragmentation of the system in such a way as to focus on an active site in a specific portion of the protein”, says Luigi Genovese from CEA (Commissariat à l’énergie atomique et aux énergies alternatives) who is heading Max’s efforts on this topic. “We think this modelling approach could inform efforts in protein design by granting access to variables otherwise impervious to observation.”

 The project E-CAM supports HPC simulations in industry and academia through software development, training and discussion in simulation and modeling. Project members are currently following two approaches to add to the research on the corona virus.
The project E-CAM supports HPC simulations in industry and academia through software development, training and discussion in simulation and modeling. Project members are currently following two approaches to add to the research on the corona virus.
Firstly, the SARS-CoV-2 virus that causes COVID-19 uses a main protease to be functional. One of the drug targets currently under investigation is an inhibitor for this protease. While efforts on simulations of binding stability and dynamics are being conducted, not much is known of the dynamical transitions of the binding-unbinding reaction. Yet, this knowledge is crucial for improved drug design. E-CAM aims to shed light on these transitions, using a software package developed by project teams at the University of Amsterdam and the Ecole Normale Superieure in Lyon.
Secondly, E-CAM contributes to the development of the software required to design a protein-based sensor for the quick detection of COVID-19. The sensor, developed at the partner University College Dublin with the initial purpose to target influenza, is now being adapted to SARS-CoV-2. This adaptation needs DNA sequences as an input for suitable antibody-epitope pairs. High-performance computing is required to identify these DNA sequences to design and simulate the sensors prior to their expression in cell lines, purification and validation.
 The project PerMedCoE aims to optimise codes for cell-level simulations in high-performance computing, and to bridge the gap between organ and molecular simulations. The project started in October 2020.
The project PerMedCoE aims to optimise codes for cell-level simulations in high-performance computing, and to bridge the gap between organ and molecular simulations. The project started in October 2020.
“Multiscale modelling frameworks prove useful in integrating mechanisms that have very different time and space scales, as in the study of viral infection, human host cell demise and immune cells response. Our goal is to provide such a multiscale modelling framework that includes infection mechanisms, virus propagation and detailed signalling pathways,” says Alfonso Valencia, PerMedCoE project coordinator at the Barcelona Supercomputing Center.
The project team has developed a use case that focusses on studying COVID-19 infections using single-cell data. The work was presented to the research community at a specialized virtual conference in November, the Disease Map Community Meeting. “This use case is a priority in the first months of the project”, says Valencia.
On the technical level, disease maps networks will be converted to models of COVID-19 and human cells from the lung epithelium and the immune system. Then, the team will use omics data to personalise models of different patients’ groups, differentiated for example by age or gender. These data-tailored models will then be incorporated into a COVID-focussed version of the open source cell-level simulator PhysiCell.
 The HiDALGO project focusses on modelling and simulating the complex processes which arise in connection with major global challenges. The researchers have developed the Flu and Coronavirus Simulator (FACS) with the objective to support decision makers to provide an appropriate response to the current pandemic situation taking into account health and care capabilities.
The HiDALGO project focusses on modelling and simulating the complex processes which arise in connection with major global challenges. The researchers have developed the Flu and Coronavirus Simulator (FACS) with the objective to support decision makers to provide an appropriate response to the current pandemic situation taking into account health and care capabilities.
FACS is guided by the outcomes of SEIR (Susceptible-Exposed-Infectious-Recovered) models operating at national level. It uses geospatial data sources from Openstreet Map to approximate the viral spread in crowded places, while trading the potential routes to reach them.
In this way, the simulator can model the COVID-19 spread at local level to provide estimations of infections and hospital arrivals, given a range of public health interventions, going from no interventions to lockdowns. Public authorities can use the results of the simulations to identify peaks of contagion, set appropriate measures to reduce spread and provide necessary means to hospitals to prevent collapses. “FACS has enabled us to forecast the spread of COVID-19 in regions such as the London Borough of Brent. These forecasts have helped local National Health Service Trusts to more effectively plan out health and care services in response to the pandemic.” says Derek Groen from the HiDALGO project partner Brunel University London.

![]() EXCELLERAT is a project that is usually focussing on supercomputing applications in the area of engineering. Nevertheless, a group of researchers from EXCELLERAT’s consortium partner SSC-Services GmbH, an IT service provider in Böblingen, Germany and the High-Performance Computing Center Stuttgart (HLRS) are also providing measures to contain the pandemic by supporting the German Federal Institute for Population Research (Bundesinstitut für Bevölkerungsforschung, BiB).
EXCELLERAT is a project that is usually focussing on supercomputing applications in the area of engineering. Nevertheless, a group of researchers from EXCELLERAT’s consortium partner SSC-Services GmbH, an IT service provider in Böblingen, Germany and the High-Performance Computing Center Stuttgart (HLRS) are also providing measures to contain the pandemic by supporting the German Federal Institute for Population Research (Bundesinstitut für Bevölkerungsforschung, BiB).
The scientists have developed an intelligent data transfer platform, which enables the BiB to upload data, perform computing-heavy simulations on the HLRS’ supercomputer Hawk, and download the results. The platform supports the work of BiB researchers in predicting the demand for intensive care units during the COVID-19 pandemic. “Nowadays, organisations face various issues while dealing with HPC calculations, HPC in general or even the access to HPC resources,” said Janik Schüssler, project manager at SSC Services. “In many cases, calculations are too complex and users do not have the required know-how with HPC technologies. This is the challenge that we have taken on. The BiB’s researchers had to access HLRS’s Hawk in a very complex way. With the help of our new platform, they can easily access Hawk from anywhere and run their simulations remotely.”
“This platform is part of EXCELLERAT’s overall strategy and tools development, which not only addresses the simulation part of engineering workflows, but provides users the necessary means to optimise their work”, said Bastian Koller, Project Coordinator of EXCELLERAT and HLRS’s Managing Director. “Extending the applicability of this platform to further use cases outside of the engineering domain is a huge benefit and increases the impact of the work performed in EXCELLERAT.”